.svg)




What lifespan and healthspan actually measure
Lifespan definition is straightforward: it's the total number of years a person lives from birth to death. When we talk about life expectancy rising globally, we're talking about lifespan (WHO on aging and health). It's a simple count of years, regardless of what those years look like.
Healthspan is more nuanced. It refers to the period of life spent in good health, free from chronic disease and disability that significantly impairs function or quality of life. It's not just about being alive. It's about being able to move without pain, think clearly, maintain independence, and engage meaningfully with the world around you. Healthspan ends when chronic conditions begin to limit daily activities or when disease burden becomes substantial.
The distinction matters because medical advances have dramatically extended lifespan without proportionally extending healthspan. We've become excellent at keeping people alive longer through pharmaceuticals, surgical interventions, and intensive care. But we haven't been as successful at preventing the accumulation of cellular damage, metabolic dysfunction, and chronic inflammation that drive age-related disease. The result is that people are living more years, but spending more of those years managing multiple chronic conditions (Nature Aging: new medicines to close the healthspan-lifespan gap).
How the hallmarks of aging drive the healthspan-lifespan gap
The widening gap between healthspan and lifespan isn't random. It reflects the progressive accumulation of damage across multiple biological systems, captured in what researchers call the hallmarks of aging. These interconnected processes explain why chronic disease burden increases with age and why interventions that target one hallmark often influence others.
Cellular senescence and chronic inflammation
Senescent cells stop dividing but don't die. Instead, they secrete inflammatory molecules that damage surrounding tissue. This process, called inflammaging, accelerates when the immune system's ability to clear senescent cells declines. The inflammatory signaling drives insulin resistance, atherosclerosis, and neurodegeneration. It's a major reason why metabolic and cardiovascular diseases cluster in older adults.
Mitochondrial dysfunction and metabolic decline
Mitochondria generate cellular energy and regulate metabolic signaling. As mitochondrial function declines with age, cells produce less ATP and more reactive oxygen species. This impairs tissue repair, reduces exercise capacity, and contributes to sarcopenia. Mitochondrial dysfunction also disrupts nutrient sensing pathways like mTOR and AMPK, which regulate how cells respond to food intake and energy availability.
Loss of proteostasis and protein aggregation
Cells maintain protein quality through synthesis, folding, and degradation. When this system fails, misfolded proteins accumulate and form aggregates that are toxic to neurons and other cells. This process underlies Alzheimer's disease, Parkinson's disease, and other neurodegenerative conditions that disproportionately affect healthspan in the final years of life.
Epigenetic alterations and biological age acceleration
Epigenetic marks regulate which genes are active without changing DNA sequence. These marks drift with age, silencing genes needed for cellular repair and activating inflammatory pathways. Epigenetic clocks measure this drift and predict disease risk independent of chronological age. Accelerated epigenetic aging is associated with shorter healthspan even when lifespan is normal.
What drives the gap between healthspan and lifespan
The healthspan-lifespan gap doesn't widen uniformly. Specific behavioral, metabolic, and environmental factors accelerate biological aging and determine how many years are spent in poor health. Understanding these drivers provides insight into which systems require attention to compress morbidity and extend years lived in good health.
Metabolic dysfunction and insulin resistance
Chronic hyperglycemia and hyperinsulinemia drive advanced glycation end-product formation, which cross-links proteins and impairs tissue function. Insulin resistance disrupts cellular nutrient sensing, promoting fat accumulation in liver and muscle while impairing mitochondrial function. This cascade underlies type 2 diabetes, cardiovascular disease, and fatty liver disease. Tracking fasting insulin, HbA1c, and triglyceride-glucose index provides early signals of metabolic aging before overt disease appears.
Chronic inflammation from multiple sources
Systemic inflammation accelerates aging through multiple mechanisms. Visceral fat secretes inflammatory cytokines that promote insulin resistance and atherosclerosis. Gut dysbiosis allows bacterial endotoxins to enter circulation, triggering immune activation. Chronic stress elevates cortisol, which suppresses immune function while promoting inflammatory signaling in other tissues. High-sensitivity CRP and inflammatory ratios like neutrophil-to-lymphocyte ratio capture this systemic inflammatory burden.
Sedentary behavior and muscle loss
Physical inactivity accelerates sarcopenia, the age-related loss of muscle mass and strength. Muscle is metabolically active tissue that regulates glucose disposal, produces anti-inflammatory myokines, and supports mitochondrial biogenesis. Loss of muscle mass predicts disability, falls, and mortality more strongly than most blood biomarkers. Resistance training stimulates mTOR-mediated protein synthesis, while aerobic exercise activates PGC-1alpha to generate new mitochondria.
Sleep disruption and circadian misalignment
Deep sleep is when the brain clears metabolic waste through the glymphatic system and when growth hormone secretion peaks to support tissue repair. Chronic sleep deprivation accelerates epigenetic aging, impairs glucose metabolism, and increases inflammatory markers. Circadian rhythm disruption from shift work or irregular sleep schedules compounds these effects by desynchronizing metabolic and immune function.
Environmental toxin exposure
Accumulated exposure to heavy metals, air pollution, and endocrine-disrupting chemicals contributes to the healthspan-lifespan gap. These toxins generate oxidative stress, impair mitochondrial function, and disrupt hormonal signaling. The Environmental Toxin Panel measures urinary metabolites of pesticides, plasticizers, and industrial chemicals that accumulate over decades.
Why the gap varies so dramatically between individuals
Two people born in the same year can have radically different healthspan trajectories. One may remain functionally independent into their nineties while another develops multiple chronic conditions in their sixties. This variation reflects differences in genetic architecture, metabolic baseline, and cumulative exposure history.
APOE4 genotype increases Alzheimer's risk and cardiovascular disease susceptibility. FOXO3 variants are overrepresented in centenarians and associated with enhanced stress resistance. Genetic determinants of telomere length influence the rate of cellular aging, though telomere length itself is a noisy biomarker with high individual variability. These genetic factors set a baseline, but lifestyle and environment determine how that genetic potential is expressed.
Biological age measured by epigenetic clocks like GrimAge and DunedinPACE diverges significantly from chronological age. Some individuals age epigenetically faster due to chronic stress, poor metabolic health, or smoking history. Others age more slowly through consistent exercise, caloric moderation, and low inflammatory burden. The pace of epigenetic aging predicts disease onset and mortality independent of traditional risk factors.
Individual variation in insulin sensitivity, mitochondrial efficiency, and fat oxidation capacity affects how the body responds to dietary patterns and exercise. Some people maintain excellent glucose control and low inflammatory markers despite modest physical activity, while others develop insulin resistance despite regular exercise. This metabolic heterogeneity explains why the same intervention produces different healthspan outcomes across individuals.
Women experience a 2.4-year larger healthspan-lifespan gap than men globally. Menopause accelerates bone loss, increases cardiovascular risk, and shifts body composition toward visceral fat accumulation. The loss of estrogen's protective effects on vascular function and metabolic regulation compresses healthspan in the final decades. Men experience a more gradual decline in testosterone, which affects muscle mass, bone density, and metabolic resilience.
Early life adversity, chronic psychological stress, and repeated exposure to discrimination or financial instability accumulate as allostatic load. This cumulative burden accelerates telomere shortening, elevates baseline cortisol, and increases systemic inflammation. Individuals with high allostatic load enter older age with depleted physiological reserves, narrowing the margin between healthspan and lifespan.
What the evidence actually shows about closing the gap
The research on extending healthspan is more robust than the research on extending lifespan in humans. We have strong evidence for interventions that compress morbidity, meaning they delay disease onset and reduce the number of years spent in poor health.
Caloric restriction extends lifespan in yeast, worms, flies, and rodents through mechanisms involving AMPK activation, mTOR inhibition, and enhanced autophagy. The CALERIE trial in humans demonstrated that moderate caloric restriction improves metabolic markers, reduces oxidative stress, and slows biological aging measured by epigenetic clocks. However, long-term effects on human lifespan remain unknown, and the intervention is difficult to sustain.
VO2 max, a measure of aerobic capacity, is one of the strongest predictors of all-cause mortality. Each metabolic equivalent increase in VO2 max reduces mortality risk by approximately 13%. Resistance training preserves muscle mass and bone density, both critical for maintaining independence in later life. The evidence here is unambiguous: regular physical activity extends healthspan through multiple mechanisms including mitochondrial biogenesis, improved insulin sensitivity, and anti-inflammatory myokine secretion.
Metformin activates AMPK and improves insulin sensitivity. Observational data suggests it may reduce cancer risk and slow aging, but the TAME trial testing metformin for longevity in humans is ongoing. Rapamycin inhibits mTOR and extends lifespan in mice, but human data is limited to small studies in specific disease populations. NAD+ precursors like NMN and NR have strong mechanistic rationale and improve metabolic markers in animal models, but human RCT evidence on longevity outcomes is still preliminary.
Senolytics like dasatinib and quercetin selectively eliminate senescent cells in animal models, improving physical function and reducing age-related pathology. Early human trials show promise in osteoarthritis and idiopathic pulmonary fibrosis, but whether senolytics extend healthspan in otherwise healthy aging populations is unproven. The field is moving quickly, but clinical translation lags behind the hype.
The strongest evidence supports using biomarkers to identify and address modifiable risk factors early. Lowering ApoB reduces cardiovascular events. Improving fasting insulin and HbA1c prevents progression to diabetes. Reducing hsCRP through weight loss, exercise, or anti-inflammatory interventions lowers systemic inflammatory burden. These interventions don't just extend lifespan. They compress the years spent managing chronic disease.
Measuring what actually matters for your healthspan
Closing the healthspan-lifespan gap in your own life requires knowing where you stand biologically, not just chronologically. A single measurement is a snapshot. A series of measurements over time reveals trajectory and rate of change, which matter more than any single data point.
Metabolic markers like fasting insulin, HbA1c, and triglyceride-glucose index capture insulin sensitivity and glucose regulation before overt diabetes develops. Cardiovascular markers including ApoB, Lp(a), and hsCRP identify atherosclerotic risk decades before symptoms appear. Inflammatory markers like hsCRP and inflammatory ratios reflect systemic inflammatory burden that accelerates multiple aging pathways simultaneously.
Hormonal markers including IGF-1, DHEA-S, testosterone, and thyroid function provide insight into anabolic capacity and metabolic rate. Nutrient status markers like vitamin D, magnesium identify deficiencies that impair cellular function and accelerate aging.
Body composition measured by DEXA scan reveals lean mass, fat mass, and visceral fat area. Muscle mass is one of the strongest predictors of long-term metabolic and physical resilience. Grip strength, though not a blood test, functions as a biomarker of overall physiological reserve and predicts disability and mortality.
Tracking your biological age over time
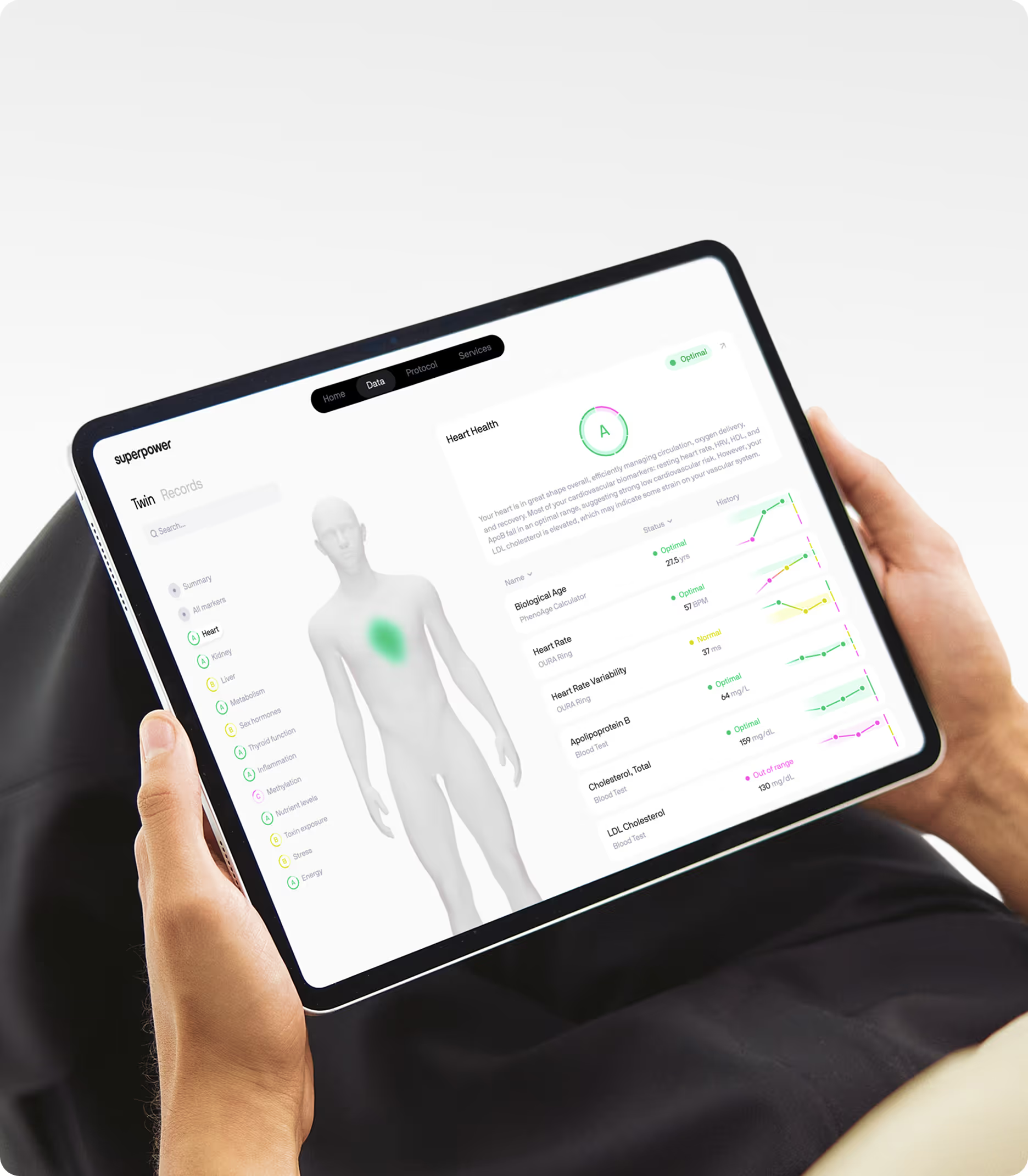
If you want to know whether you're closing or widening your personal healthspan-lifespan gap, you need comprehensive baseline data across the metabolic, inflammatory, and hormonal systems most relevant to how well and how long you live. Standard annual bloodwork typically measures cholesterol and glucose but misses the markers that predict biological aging and chronic disease risk years in advance. Superpower's 100+ biomarker panel covers fasting insulin, ApoB, Lp(a), hsCRP, homocysteine, and the nutrient and hormonal markers that reveal whether your biology is aging faster or slower than your chronological age. Tracking these markers longitudinally gives you the data to make informed decisions about interventions that extend healthspan, not just lifespan.



FAQs
Lifespan counts total years lived from birth to death. Healthspan refers to the years spent in good health — free from chronic disease or disability that significantly limits function or quality of life. Medical advances have extended lifespan through pharmaceuticals and intensive care, but have been less effective at preventing the cellular damage, metabolic dysfunction, and inflammation that drive age-related disease. The result is more years lived, but more of those years spent managing chronic conditions.
As of 2019, the global gap between healthspan and lifespan had widened to 9.6 years, meaning on average people spend nearly a decade at the end of life in poor health. In the United States specifically, Americans now spend an average of 12.4 years in poor health — well above the global average. Women experience a 2.4-year larger gap than men across 183 countries, largely because of the metabolic and cardiovascular consequences of menopause.
The widening gap reflects progressive accumulation of damage across multiple biological systems known as the hallmarks of aging. Senescent cells secrete inflammatory molecules that drive atherosclerosis and insulin resistance. Mitochondrial dysfunction reduces ATP production and increases oxidative stress. Loss of proteostasis allows misfolded proteins to aggregate, causing neurodegeneration. Epigenetic drift silences repair genes and activates inflammatory pathways. These interconnected processes accelerate chronic disease onset while lifespan continues to extend.
Chronic hyperglycemia and hyperinsulinemia drive formation of advanced glycation end products, which cross-link proteins and impair tissue function throughout the body. Insulin resistance disrupts cellular nutrient sensing, promotes fat accumulation in the liver and muscle, and impairs mitochondrial function. This cascade underlies type 2 diabetes, cardiovascular disease, and fatty liver disease. Fasting insulin and HbA1c can detect metabolic dysfunction years before overt disease appears, creating a window for intervention.
Women experience a 2.4-year larger healthspan-lifespan gap than men globally. Menopause accelerates bone loss, increases cardiovascular risk, and shifts body composition toward visceral fat accumulation. The loss of estrogen's protective effects on vascular function and metabolic regulation compresses healthspan in the final decades of life. Men experience a more gradual hormonal decline, with testosterone decreasing steadily over decades rather than abruptly, which affects muscle mass, bone density, and metabolic resilience differently.
The strongest evidence supports physical activity and metabolic biomarker tracking as tools for extending healthspan. Each MET increase in VO2 max reduces mortality risk by approximately 13%. The CALERIE trial demonstrated that moderate caloric restriction improved metabolic markers and slowed biological aging measured by epigenetic clocks. Lowering ApoB reduces cardiovascular events; improving fasting insulin and HbA1c prevents diabetes progression. These interventions compress the years spent managing chronic disease rather than simply extending lifespan.
References
- Mayoclinic. (n.d.). Mayo clinic q and a lifespan vs healthspan. https://newsnetwork.mayoclinic.org/discussion/mayo-clinic-q-and-a-lifespan-vs-healthspan
- Crimmins, E. M. (2015). Lifespan and Healthspan: Past, Present, and Promise. The Gerontologist, 55(6), 901-11. https://doi.org/10.1093/geront/gnv130
- The Lancet. (n.d.). Lancet. https://thelancet.com/journals/lancet/article/PIIS0140-6736(20)30925-9/fulltext
- Healthdata. (n.d.). Global Burden of Disease (GBD). https://healthdata.org/research-analysis/gbd
- National Institute on Aging. (n.d.). About NIA. https://nia.nih.gov/about/aging-strategic-directions-research/goal-biology-impact
- World Health Organization. (n.d.). Ageing and health. https://who.int/news-room/fact-sheets/detail/ageing-and-health
- Crane, P. A., Wilkinson, G., & Teare, H. (2022). Healthspan versus lifespan: new medicines to close the gap. Nature aging, 2(11), 984-988. https://doi.org/10.1038/s43587-022-00318-5

























.avif)
.svg)
.svg)


