.svg)




LDL cholesterol, defined as a cholesterol mass
LDL cholesterol (LDL-C) estimates how much cholesterol is carried inside low-density lipoprotein particles — it measures the cargo, not the number of vehicles. Each LDL particle carries cholesterol, phospholipids, and a structural protein called apolipoprotein B (ApoB). LDL-C tells you how much cholesterol is riding along; ApoB and LDL-P tell you how many particles are on the road. Both answer different questions about cardiovascular traffic.
Why LDL-C is a mass, not a count
Your liver packages triglycerides and cholesterol into lipoproteins and sends them into circulation. LDL forms as larger VLDL particles deliver triglycerides to tissues and shrink down. LDL's role is to deliver cholesterol for cell membranes, hormone synthesis, and repair.
LDL particles drift in the bloodstream and, over time, some slip into the artery wall. If too many particles linger — especially in an inflamed environment — they can become modified and trigger an immune response. Foam cells form, plaque builds, and the artery gradually narrows. It is a slow, cumulative process influenced by particle number, time, and vessel biology. Because LDL-C measures cholesterol mass rather than particle number, it does not directly count how many particles are interacting with artery walls — ApoB does that.
The scale of this accumulation matters for long-term health. Mendelian randomization studies and large randomized trials converge: for every ~39 mg/dL (1 mmol/L) reduction in LDL-C, major cardiovascular events drop by roughly 20–25%, with larger absolute benefits the longer and earlier the reduction occurs.
Reading low, normal, and high LDL-C
Reference intervals on a lab report reflect where most people fall, not necessarily what is best for long-term artery health. "Normal" is statistical; risk-based targets are more specific to individual context.
Decades of trials and genetic studies point in the same direction: lower lifetime LDL-C exposure is linked to lower cardiovascular risk. Many guidelines use <100 mg/dL as an optimal general target and <70 mg/dL for people at higher cardiovascular risk. The specifics vary by country, clinical society, and individual history, which is why two clinicians can reasonably emphasize different cut points while both following the evidence.
Ranges are also lab-specific. LDL-C is most often calculated from total cholesterol, HDL-C, and triglycerides using the Friedewald equation, which can be less accurate when triglycerides are high or in nonfasting states. Direct LDL-C assays and newer equations can help, but they are not interchangeable — two labs can return slightly different numbers on the same day. Age, sex, and life stage matter too: LDL tends to run lower in many premenopausal women and rises around menopause; pregnancy naturally shifts lipids upward; children and teens have different norms.
When levels run high
An elevated LDL-C often reflects either increased lipoprotein production by the liver or reduced clearance through LDL receptors. Common physiological drivers include high intake of saturated fats, which reduces LDL receptor activity so LDL lingers longer in circulation; lower thyroid function; and weight gain with greater VLDL output. Genetics also play a major role — in familial hypercholesterolemia (FH), LDL receptors do not work well, so LDL stays high from a young age.
Context sharpens the picture. If triglycerides are high and HDL-C is low, the liver may be overproducing VLDL, generating more LDL downstream. If ApoB is elevated, more atherogenic particles are on the road. If Lp(a) is high, its cholesterol content is included in the calculated LDL-C figure, which can inflate the result and make ApoB a cleaner read of true particle burden. A solitary LDL-C spike during illness or rapid weight loss may be transient; persistence across repeat tests is more telling.
When levels run low
Low LDL-C can reflect efficient clearance, lower particle production, or genetic variants that naturally favor low LDL — some people inherit variants that mimic the effect of modern lipid-lowering therapies and live with very low LDL lifelong without apparent harm.
Unexpectedly low LDL-C can also appear with hyperthyroidism, malnutrition, advanced liver disease, severe inflammation, or some cancers, so low is not automatically favorable — it is a data point that deserves context. Assay nuance matters here as well: if Lp(a) is high, it can inflate calculated LDL-C, meaning a result that looks normal may mask a higher true LDL-C once Lp(a) is accounted for. In discordant cases — for example, LDL-C looks unremarkable but ApoB is elevated — particle count often tracks risk more closely than cargo alone.
What shifts LDL-C independent of dietary fat
Several factors can move LDL-C in ways that are worth understanding when interpreting a result.
- Saturated fat intake reduces LDL receptor activity in the liver, so LDL particles remain in circulation longer and calculated LDL-C rises. Swapping some saturated fats for unsaturated fats nudges receptors to clear LDL more efficiently.
- Weight loss can transiently raise LDL-C as stored cholesterol mobilizes out of tissues, even while ApoB and triglycerides improve. Trends over months reflect the true direction of change.
- Sleep deprivation and chronic stress tilt hormones toward higher cortisol and catecholamines, increasing hepatic VLDL output and downstream LDL formation.
- High triglycerides or nonfasting draws reduce the accuracy of the Friedewald equation used to calculate LDL-C; when triglycerides exceed 400 mg/dL, the calculated value becomes unreliable and a direct assay is preferable.
- Lp(a) is genetically determined and its cholesterol content is counted inside calculated LDL-C, meaning elevated Lp(a) can inflate the LDL-C figure independent of LDL particle activity.
- Thyroid status, kidney health, liver function, and pregnancy all influence LDL-C interpretation and should be considered when results are unexpected.
- Lipid-lowering medications — statins, ezetimibe, and PCSK9 inhibitors — work by different mechanisms (boosting LDL receptor activity, reducing cholesterol synthesis, blocking absorption, or enhancing clearance) and produce meaningful, measurable shifts in LDL-C.
The markers that put LDL-C in cardiovascular context
LDL-C is part of a broader particle family. Interpreting it alongside related markers resolves the cargo-vs.-count ambiguity and surfaces risk that LDL-C alone can miss.
- ApoB — ApoB counts every atherogenic particle: each LDL, VLDL remnant, and Lp(a) carries exactly one ApoB molecule. When LDL-C and ApoB disagree, risk tracks the particle count. High ApoB with normal LDL-C is common in insulin resistance, where particles carry less cholesterol each — a pattern LDL-C alone would miss.
- LDL-P — LDL particle number directly quantifies the traffic of LDL particles interacting with artery walls. LDL-P and LDL-C can diverge significantly in metabolic syndrome, making LDL-P the more reliable risk signal when the two are discordant.
- Lp(a) — Lp(a) is genetically determined and its cholesterol is counted inside calculated LDL-C. High Lp(a) can inflate LDL-C, making ApoB a cleaner read of atherogenic particle burden when Lp(a) is elevated.
- Triglycerides — elevated triglycerides reduce LDL-C calculation accuracy via the Friedewald equation and signal higher VLDL output that generates more LDL downstream. Triglycerides contextualize the LDL-C value, particularly in nonfasting states.
- Non-HDL cholesterol — Non-HDL cholesterol (total cholesterol minus HDL-C) captures all atherogenic cholesterol in a single figure and performs better than LDL-C in nonfasting states and when triglycerides are elevated.
Seen together, these markers map a network. High ApoB with normal LDL-C points to particle-heavy traffic with lighter cargo. High non-HDL-C and triglycerides suggest the liver may be overproducing VLDL. Elevated Lp(a) with modest LDL-C means baseline risk is higher than LDL-C alone suggests.
When to retest LDL-C after a diet or statin change
LDL-C responds to interventions on a predictable timeline. Statin-driven reductions reach steady state at roughly 4–6 weeks; dietary saturated-fat changes typically settle by 8–12 weeks. Retesting before those windows close captures a moving target rather than a stable value.
As a general guide: retest every 3 months when actively adjusting diet, medication, or lifestyle; retest annually during maintenance once results are stable.
Draw conditions matter for accuracy. A fasting draw is preferred when triglycerides may be elevated, since the Friedewald equation becomes less reliable in nonfasting states and at triglyceride levels above 400 mg/dL. Using a consistent morning protocol — same lab, same fasting window — removes variables that can make two results look different when the underlying biology has not changed. Different calculation methods and labs can produce slightly different values on the same day; comparing across labs adds noise to trend interpretation.
When your LDL-C deserves a cardiovascular workup
Testing turns guesswork into trendlines. A persistently elevated LDL-C — especially when accompanied by high ApoB, elevated non-HDL-C, or a family history of early heart disease — warrants a fuller cardiovascular assessment rather than a single-number response. If results drift in the wrong direction across repeat tests, that pattern is an early warning with a clear place to look.
Track how you feel and perform alongside the numbers. If training improves, sleep steadies, and ApoB falls, that is a coherent signal. If LDL-C remains high despite dietary changes and other risk factors are stacking up, partnering with a clinician to personalize targets is the appropriate next step.
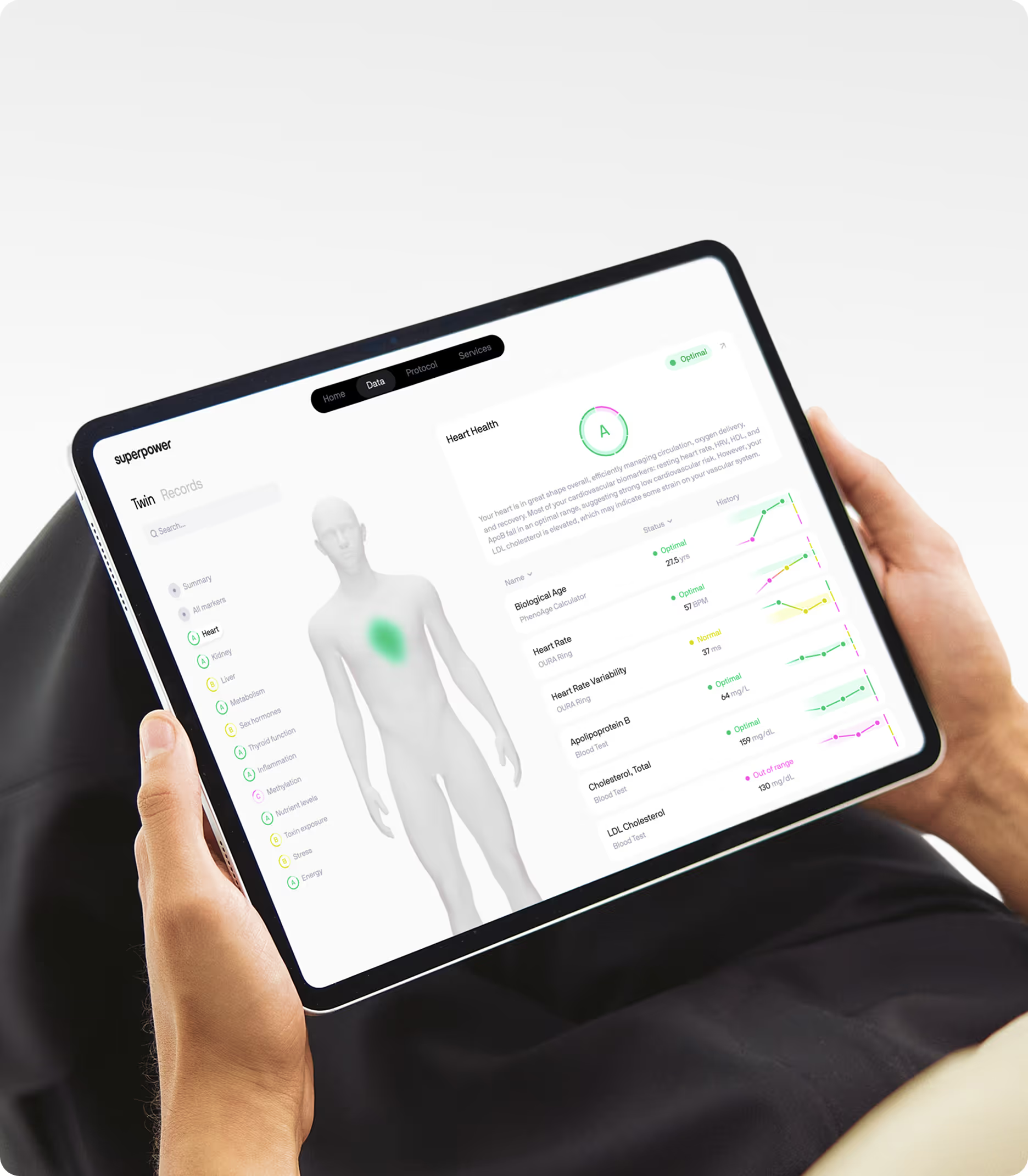
At Superpower, the approach is to measure LDL-C alongside ApoB, LDL-P, non-HDL cholesterol, triglycerides, and Lp(a) so you stop playing the one-number game. That is how you move from a snapshot to a trend, and from generic advice to a plan grounded in your own biology. Learn more about the approach.



FAQs
References
- Ference, B. A., Ginsberg, H. N., Graham, I., Ray, K. K., Packard, C. J., Bruckert, E., Hegele, R. A., Krauss, R. M., Raal, F. J., Schunkert, H., Watts, G. F., Borén, J., Fazio, S., Horton, J. D., Masana, L., Nicholls, S. J., Nordestgaard, B. G., van de Sluis, B., Taskinen, M. R., ... Catapano, A. L. (2017). Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. European heart journal, 38(32), 2459-2472. https://doi.org/10.1093/eurheartj/ehx144
- Silverman, M. G., Ference, B. A., Im, K., Wiviott, S. D., Giugliano, R. P., Grundy, S. M., Braunwald, E., & Sabatine, M. S. (2016). Association Between Lowering LDL-C and Cardiovascular Risk Reduction Among Different Therapeutic Interventions: A Systematic Review and Meta-analysis. JAMA, 316(12), 1289-97. https://doi.org/10.1001/jama.2016.13985
- Wang, N., Fulcher, J., Abeysuriya, N., Park, L., Kumar, S., Di Tanna, G. L., Wilcox, I., Keech, A., Rodgers, A., & Lal, S. (2020). Intensive LDL cholesterol-lowering treatment beyond current recommendations for the prevention of major vascular events: a systematic review and meta-analysis of randomised trials including 327 037 participants. The lancet. Diabetes & endocrinology, 8(1), 36-49. https://doi.org/10.1016/S2213-8587(19)30388-2
- Johannesen, C. D. L., Mortensen, M. B., Langsted, A., & Nordestgaard, B. G. (2021). Apolipoprotein B and Non-HDL Cholesterol Better Reflect Residual Risk Than LDL Cholesterol in Statin-Treated Patients. Journal of the American College of Cardiology, 77(11), 1439-1450. https://doi.org/10.1016/j.jacc.2021.01.027
- Sniderman, A. D., Williams, K., Contois, J. H., Monroe, H. M., McQueen, M. J., de Graaf, J., & Furberg, C. D. (2011). A meta-analysis of low-density lipoprotein cholesterol, non-high-density lipoprotein cholesterol, and apolipoprotein B as markers of cardiovascular risk. Circulation. Cardiovascular quality and outcomes, 4(3), 337-45. https://doi.org/10.1161/CIRCOUTCOMES.110.959247
























.avif)
.svg)
.svg)


