.svg)




The atherogenic coefficient, defined in plain terms
The atherogenic coefficient (AC) is calculated as (Total cholesterol − HDL cholesterol) ÷ HDL cholesterol — equivalently, non-HDL cholesterol divided by HDL cholesterol. Non-HDL captures the cholesterol carried by particles that can promote plaque, including LDL, VLDL, IDL, and Lp(a); HDL participates in reverse cholesterol transport. The result is unitless as long as both inputs share the same unit (mg/dL or mmol/L). Mathematically, AC equals the total cholesterol-to-HDL ratio minus one. The measure has been used in epidemiologic research as a single-number expression of the balance between atherogenic delivery and protective clearance; lower AC values align with lower cardiovascular risk across those studies.
Why non-HDL versus HDL beats either alone
Total cholesterol is a blunt instrument. A person can sit at a perfectly average total cholesterol of 190 mg/dL while carrying an unfavorable mix of particles — high non-HDL, low HDL — that creates a net atherogenic environment. The ratio surfaces that imbalance where a single number cannot.
The underlying physiology is a delivery-versus-clearance story. Atherogenic particles — LDL, VLDL, IDL, remnants — deposit cholesterol at the artery wall. HDL participates in reverse cholesterol transport, shuttling cholesterol back toward the liver. When delivery outpaces clearance, the arterial environment tilts toward plaque formation. AC captures that tilt directly: the numerator represents the atherogenic load, the denominator the protective counterforce. A rising AC means the balance is shifting in the wrong direction even if neither component alone has crossed a flagged threshold.
AC shifts with real-life stressors. A few nights of poor sleep can lower HDL and raise triglyceride-rich particles, nudging AC upward. A block of consistent endurance training tends to lower non-HDL and can lift HDL over weeks, pushing AC downward. During an infection, inflammation can depress HDL function and levels, temporarily bumping AC. Diet swings matter too: refined-carb heavy meals drive up VLDL production from the liver, raising non-HDL; fiber-rich, unsaturated-fat patterns move it in the opposite direction. Single readings are snapshots; the plotline is what counts.
How the atherogenic coefficient is computed
The formula has two equivalent forms:
- AC = (Total cholesterol − HDL-C) ÷ HDL-C
- AC = non-HDL cholesterol ÷ HDL-C
Both inputs must be in the same unit — mg/dL or mmol/L — and the result is unitless. Note that AC is simply the total cholesterol-to-HDL ratio minus 1. No fasting is required for the draw: total cholesterol and HDL-C are stable without fasting. Triglycerides are the fasting-sensitive lipid on a standard panel; they are not direct inputs to AC.
Worked example
A reader has a total cholesterol of 200 mg/dL and an HDL-C of 55 mg/dL. Non-HDL cholesterol is therefore 200 − 55 = 145 mg/dL. Dividing: 145 ÷ 55 = AC 2.64 — a value in the favorable range by most epidemiologic benchmarks. That said, a companion ApoB is needed to confirm actual particle burden before drawing firm conclusions.
Reading your atherogenic coefficient score in context
There is no single universal guideline-endorsed cutoff for AC. Labs report reference intervals based on population distributions, not guaranteed protection thresholds. Epidemiologic research has generally used the following benchmarks, which can be read through two frames — a conventional frame and a more stringent preventive frame:
- AC below 3 — favorable (conventional) / target zone (preventive): Non-HDL is low relative to HDL. This range is broadly associated with lower cardiovascular risk in epidemiologic data. A preventive lens treats values well below 3 as the goal, particularly when confirmed by a low ApoB.
- AC 3–4 — moderate (conventional) / worth investigating (preventive): The balance is less favorable. In a conventional clinical context this may not trigger immediate action; a preventive approach treats this range as a prompt to examine the full lipid picture — ApoB, triglycerides, and metabolic context — before deciding whether intervention is warranted.
- AC above 4 — higher risk (conventional) / actionable (preventive): Non-HDL substantially outweighs HDL. Both frames treat this as a signal requiring clinical attention, though the threshold for intervention will depend on overall cardiovascular risk, age, sex, and comorbidities.
Important context: premenopausal women often carry higher HDL and therefore lower AC than age-matched men; menopause can narrow that gap. Pregnancy naturally lifts total cholesterol and triglycerides, which can raise AC temporarily. HDL assay methods can vary slightly across laboratory platforms, so a result near a boundary is best interpreted with that variability in mind rather than treated as a precise cutoff.
AC is not a diagnosis. Use it as a conversation starter with your clinician, layered alongside ApoB, triglycerides, Lp(a), and your overall risk profile.
What pushes the atherogenic coefficient higher or lower
AC is the ratio of two moving parts, so anything that shifts non-HDL, HDL, or both will move it. Several mechanisms are worth understanding.
VLDL overproduction and insulin resistance
The liver's output of VLDL — the triglyceride-rich precursor to LDL and remnant particles — is the primary driver of non-HDL in people with metabolic dysfunction. Insulin resistance, liver fat accumulation, and diets heavy in refined carbohydrates all amplify hepatic VLDL synthesis, raising the numerator of AC. Patterns that steady insulin and reduce liver fat — adequate protein, soluble fiber, minimizing ultra-processed foods and sugary beverages — work through this mechanism to bring non-HDL down.
HDL kinetics
HDL is the slower-moving input. Aerobic training upregulates lipoprotein lipase activity, clears triglyceride-rich lipoproteins, and over weeks to months can modestly raise HDL-C. Resistance training builds metabolically active tissue that reinforces those shifts. Expect HDL to respond on a 4–8 week timescale at minimum; non-HDL tends to respond somewhat faster to dietary change. Overtraining without adequate recovery can backfire by elevating stress hormones and disrupting sleep, which counteracts the HDL benefit.
Dietary pattern
Diets rich in unsaturated fats — fish, nuts, olive oil, avocados — tend to lower atherogenic particles compared with patterns high in refined carbohydrates and trans fats. Soluble fiber from oats, psyllium, beans, and fruit binds bile acids in the gut, prompting the liver to pull more cholesterol from circulation and nudging non-HDL downward. Marine omega-3 fatty acids limit hepatic VLDL production, which can improve non-HDL and AC. Moderate-to-high alcohol intake can raise triglycerides and push AC upward.
Sleep and stress
Short sleep and circadian misalignment increase sympathetic tone, worsen insulin resistance, and elevate triglycerides — all of which raise AC. High, sustained stress has similar effects through cortisol and inflammatory signaling. Consistent sleep timing stabilizes the hormonal rhythms that govern hepatic lipid output and HDL turnover.
Medical and physiological factors
Statins reduce LDL and non-HDL by inhibiting hepatic cholesterol synthesis. Ezetimibe lowers intestinal cholesterol absorption. PCSK9 inhibitors cut LDL substantially by increasing LDL receptor recycling. Fibrates and high-dose omega-3s lower triglycerides, reducing remnant lipoproteins. GLP-1–based therapies that reduce weight and improve insulin sensitivity can indirectly improve AC through broad metabolic effects. Thyroid function is pivotal: hypothyroidism raises LDL and non-HDL, while hyperthyroidism can lower them. Kidney disease, liver disease, and nephrotic syndrome alter lipid trafficking. Combined hormonal contraceptives and pregnancy elevate triglycerides; menopause can lower HDL and increase LDL.
Markers that surround the atherogenic coefficient
- HDL cholesterol — the denominator of AC; a rising HDL moves the ratio favorably independent of any change in total cholesterol.
- Total cholesterol — the parent value from which the numerator is derived; subtracting HDL-C from total cholesterol gives non-HDL, the atherogenic load in the ratio.
- ApoB — counts atherogenic particles directly; two people can have identical AC with meaningfully different ApoB and therefore different cardiovascular risk, making it the essential companion for confirming particle burden.
- Triglycerides — when elevated, they inflate non-HDL and push AC upward; high triglycerides also flag excess VLDL and remnant lipoprotein contribution that AC alone does not distinguish.
- Lp(a) — a genetically determined atherogenic particle that adds significant cardiovascular risk independent of AC; its contribution is not captured by the ratio.
The right retest window for the atherogenic coefficient
Both total cholesterol and HDL-C respond to lifestyle and medication changes, but at different speeds. Dietary shifts and statin initiation can move non-HDL within 4–6 weeks; HDL is the slower input, typically requiring 4–8 weeks of sustained aerobic training or dietary change before a meaningful shift registers. For that reason, 8–12 weeks after a dietary or lifestyle change is the appropriate retest window — long enough to capture HDL's response, not so long that you lose the feedback signal.
For medication changes such as statin initiation, a 6–8 week draw captures most of the LDL-C response; an 8–12 week retest then confirms the full AC shift once HDL has had time to stabilize at its new level.
No fasting is required for the draw, since total cholesterol and HDL-C are not meaningfully affected by recent food intake. Using the same laboratory and the same conditions across retests is preferred — HDL assay methods can vary slightly across platforms, and consistency reduces noise when tracking a trend.
When the atherogenic coefficient becomes a clinician question
Trend your AC across seasons of life, pair it with ApoB or non-HDL, and you will see how your daily choices and health shifts land in your arteries. That feedback loop enables earlier course correction and better alignment with your goals, whether that's running a faster 10K, feeling steadier energy, or stacking the odds for healthy decades ahead.
Bring AC to a clinician when it sits persistently above 4, when it is rising across consecutive tests despite stable routines, or when it is elevated alongside other signals — high ApoB, elevated Lp(a), high triglycerides, or a strong family history of early cardiovascular disease. A modestly elevated AC with a high ApoB points toward a surplus of atherogenic particles and warrants a more active conversation. A high AC driven mostly by low HDL but normal ApoB paints a different landscape and may call for a different approach. Medications, thyroid function, metabolic health, and alcohol intake can all tilt AC upward and are worth reviewing with your clinician before acting on a single result.
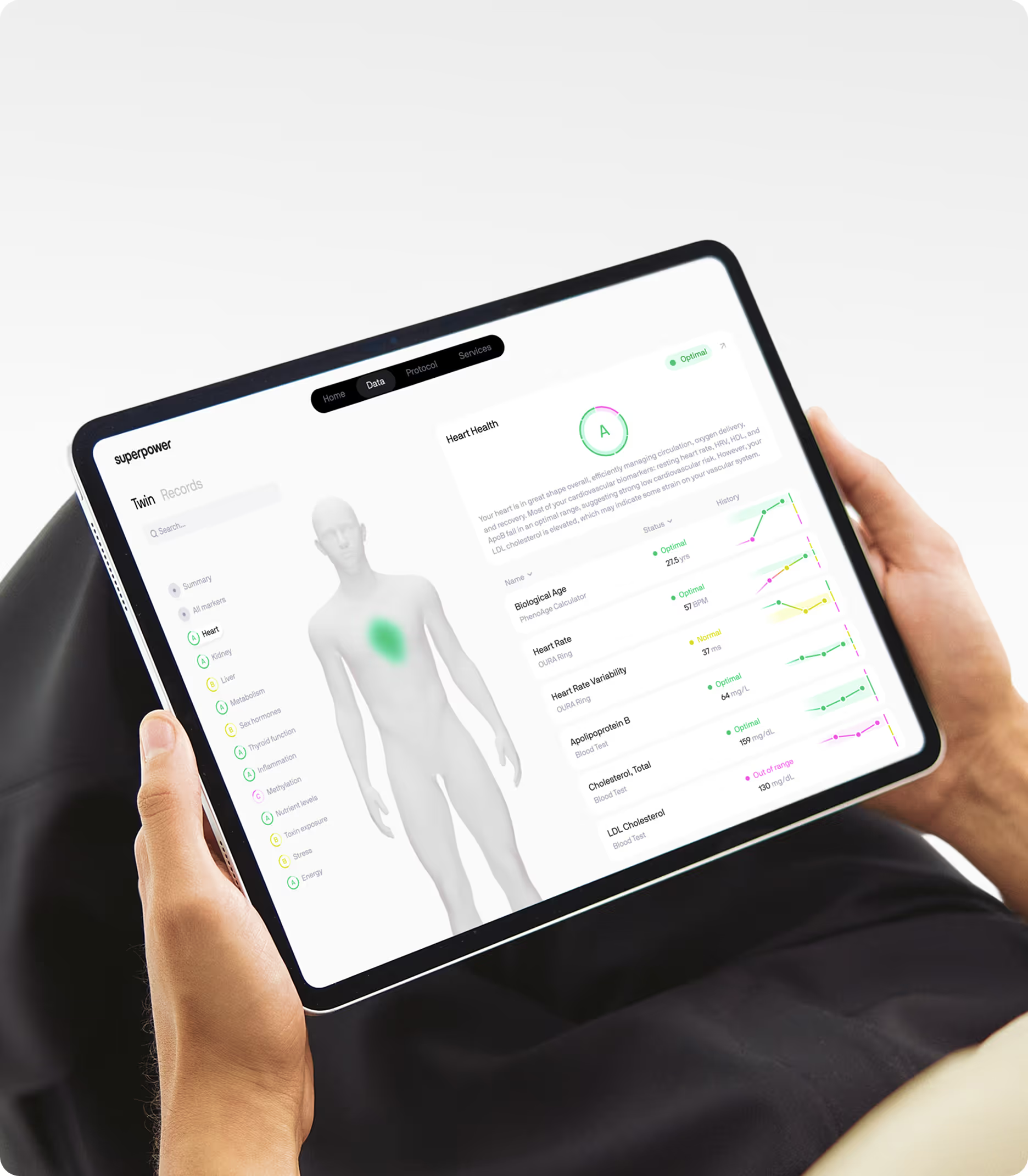
Numbers work best when they meet your lived experience. Check in with how you feel, how you train and recover, and what changes you keep. Then let the data confirm whether the physiology is moving in the direction you intend. A comprehensive panel — AC alongside ApoB, non-HDL, triglycerides, Lp(a), glucose dynamics, and inflammation — turns scattered readings into a coherent story. That is the move beyond averages, toward decisions grounded in evidence and tuned to you. Superpower is built around that approach: advanced biomarker testing with the clinical context to make sense of what you find.



FAQs
References
- Sniderman, A. D., Williams, K., Contois, J. H., Monroe, H. M., McQueen, M. J., de Graaf, J., & Furberg, C. D. (2011). A meta-analysis of low-density lipoprotein cholesterol, non-high-density lipoprotein cholesterol, and apolipoprotein B as markers of cardiovascular risk. Circulation. Cardiovascular quality and outcomes, 4(3), 337-45. https://doi.org/10.1161/CIRCOUTCOMES.110.959247
- Emerging Risk Factors Collaboration, Di Angelantonio, E., Sarwar, N., Perry, P., Kaptoge, S., Ray, K. K., Thompson, A., Wood, A. M., Lewington, S., Sattar, N., Packard, C. J., Collins, R., Thompson, S. G., & Danesh, J. (2009). Major lipids, apolipoproteins, and risk of vascular disease. JAMA, 302(18), 1993-2000. https://doi.org/10.1001/jama.2009.1619
- Sniderman, A. D., Dufresne, L., Pencina, K. M., Bilgic, S., Thanassoulis, G., & Pencina, M. J. (2024). Discordance among apoB, non-high-density lipoprotein cholesterol, and triglycerides: implications for cardiovascular prevention. European heart journal, 45(27), 2410-2418. https://doi.org/10.1093/eurheartj/ehae258
- Ference, B. A., Ginsberg, H. N., Graham, I., Ray, K. K., Packard, C. J., Bruckert, E., Hegele, R. A., Krauss, R. M., Raal, F. J., Schunkert, H., Watts, G. F., Borén, J., Fazio, S., Horton, J. D., Masana, L., Nicholls, S. J., Nordestgaard, B. G., van de Sluis, B., Taskinen, M. R., ... Catapano, A. L. (2017). Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. European heart journal, 38(32), 2459-2472. https://doi.org/10.1093/eurheartj/ehx144
- Marcovina, S. M. (2023). Lipoprotein(a): a genetically determined risk factor for Cardiovascular disease. Critical reviews in clinical laboratory sciences, 60(8), 560-572. https://doi.org/10.1080/10408363.2023.2229915
























.avif)
.svg)
.svg)


