.svg)


.svg)




What the senescence-associated secretory phenotype actually is
When cells experience severe stress from DNA damage, telomere shortening, or oncogenic signals, they can enter a state called cellular senescence. Unlike cells that undergo apoptosis and die quietly, senescent cells remain metabolically active and develop a distinct secretory profile known as the senescence-associated secretory phenotype. The SASP is a complex cocktail of pro-inflammatory cytokines, chemokines, growth factors, proteases, and extracellular matrix remodeling proteins that senescent cells continuously release into their surrounding environment.
The most prominent components include:
- Interleukin-6 (IL-6) increases markedly after DNA damage and oncogene-induced senescence across fibroblasts, keratinocytes, and epithelial cells.
- Interleukin-8 (IL-8) functions as both a cytokine and chemokine, attracting immune cells to sites of senescent cell accumulation.
- IL-1α and IL-1β contribute to the inflammatory cascade and can trigger senescence in neighboring cells.
- Tumor necrosis factor-alpha (TNF-α) amplifies systemic inflammatory responses and tissue damage.
- Monocyte chemoattractant protein-1 (MCP-1) recruits immune cells to senescent cell sites.
- Matrix metalloproteinases (MMPs) degrade extracellular matrix and disrupt tissue architecture.
- Vascular endothelial growth factor (VEGF) alters angiogenesis and vascular function.
What makes the SASP particularly problematic is its self-reinforcing nature. IL-6 and IL-8 act in an autocrine manner, binding to receptors on the same senescent cells that produced them and strengthening the senescence growth arrest. This creates a positive feedback loop where the SASP not only affects neighboring cells but also locks the senescent cell into its dysfunctional state, preventing it from either recovering or undergoing apoptosis. The composition of the SASP varies depending on the cell type, the trigger that induced senescence, and the tissue microenvironment, but the inflammatory signature remains consistent across contexts.
How the SASP connects to the hallmarks of aging
The senescence-associated secretory phenotype sits at the intersection of multiple hallmarks of aging, functioning as both a consequence of cellular damage and a driver of systemic aging processes. Cellular senescence itself is recognized as one of the primary hallmarks of aging, and the SASP is the mechanism through which senescent cells exert their most damaging effects on the organism.
Chronic inflammation and inflammaging
The SASP is the primary cellular source of chronic, low-grade inflammation that characterizes aging, a phenomenon termed inflammaging. The continuous secretion of IL-6, IL-8, IL-1α, and TNF-α by senescent cells creates a persistent inflammatory environment that elevates systemic inflammatory markers like high-sensitivity C-reactive protein. This chronic inflammation accelerates tissue dysfunction and increases susceptibility to age-related diseases including cardiovascular disease, neurodegeneration, and metabolic disorders.
Stem cell exhaustion
SASP factors directly impair stem cell function and regenerative capacity. The inflammatory cytokines and matrix metalloproteinases secreted by senescent cells disrupt stem cell niches, alter differentiation signals, and reduce the proliferative capacity of tissue-resident stem cells. This contributes to the progressive decline in tissue repair and regeneration that defines biological aging.
Altered intercellular communication
The SASP fundamentally changes how cells communicate within tissues. The secreted factors alter paracrine signaling, recruit immune cells, modify the extracellular matrix, and can even induce senescence in neighboring healthy cells through a process called secondary or bystander senescence. This spreading effect amplifies the impact of a relatively small number of senescent cells across entire tissue compartments.
Mitochondrial dysfunction
The relationship between the SASP and mitochondrial dysfunction is bidirectional (NIA: mitochondrial DNA release drives senescence and inflammation). Mitochondrial damage and increased reactive oxygen species production can trigger cellular senescence and SASP activation. Conversely, SASP factors like IL-1α can impair mitochondrial function in neighboring cells, creating a cycle of metabolic dysfunction that propagates through tissues.
What drives SASP activation and intensity
The senescence-associated secretory phenotype doesn't emerge uniformly across all senescent cells. Multiple factors determine whether a senescent cell develops a robust SASP, and the intensity and composition of the secretory profile varies based on the initiating stress and cellular context.
Key drivers include:
- Persistent DNA damage activates NF-κB signaling, which directly upregulates expression of IL-6, IL-8, and other SASP components.
- Elevated mTOR signaling drives translation of SASP factors and maintains the secretory phenotype in senescent cells.
- Cell surface-bound IL-1α functions as a master regulator by activating NF-κB and driving downstream SASP factor expression.
- Chronic psychological stress and elevated cortisol levels create a pro-inflammatory environment that enhances SASP production.
- Insulin resistance, hyperglycemia, and elevated circulating lipids enhance SASP production through increased oxidative stress and altered nutrient sensing.
The severity and persistence of DNA damage correlates with SASP intensity, explaining why exposure to ionizing radiation, chemotherapy, or chronic oxidative stress produces particularly inflammatory senescent cells. Inhibiting mTOR with rapamycin or its analogs reduces SASP production without eliminating senescent cells, demonstrating that the growth arrest and secretory phenotype are mechanistically separable. Conversely, caloric restriction and interventions that improve metabolic health like metformin can reduce SASP intensity by modulating AMPK and mTOR signaling.
Why SASP responses vary between individuals
Two people of the same chronological age can have dramatically different burdens of senescent cells and SASP-driven inflammation. This variation reflects differences in genetics, accumulated exposures, immune function, and metabolic health that determine both the rate of senescent cell accumulation and the body's capacity to clear them.
Individual variation stems from:
- Immune system competence determines how efficiently natural killer cells and macrophages eliminate senescent cells before they accumulate.
- Genetic polymorphisms in IL-6, TNF-α, and NF-κB pathway components influence baseline inflammatory tone and SASP magnitude.
- Cumulative exposure to DNA-damaging agents like UV radiation, air pollution, or tobacco smoke accelerates senescent cell accumulation.
- Insulin resistance and elevated fasting insulin levels enhance SASP production through increased oxidative stress.
- Sex hormones modulate SASP intensity, with estrogen suppressing inflammation and its loss after menopause accelerating biological aging.
With aging, immune function declines in a process called immunosenescence, reducing the efficiency of senescent cell clearance. Individuals with more robust immune function maintain lower senescent cell burdens and experience less SASP-driven inflammation. The relationship between metabolic health and SASP is bidirectional: SASP factors like IL-6 can induce insulin resistance in surrounding tissues, creating a vicious cycle where metabolic dysfunction and cellular senescence reinforce each other. Similarly, declining testosterone in aging men may reduce the body's capacity to manage senescent cell accumulation.
What the evidence actually shows about SASP and aging
The connection between the senescence-associated secretory phenotype and aging is supported by multiple lines of evidence, though the strength of data varies across different aspects of the SASP hypothesis.
The most robust evidence comes from preclinical studies in mice. Genetic elimination of senescent cells using senolytic approaches extends both healthspan and lifespan in multiple mouse models. These interventions reduce systemic inflammation, improve physical function, and delay onset of age-related pathologies including cardiovascular disease, neurodegeneration, and metabolic dysfunction. The benefits are directly attributable to removing SASP-producing cells, as interventions that suppress SASP production without eliminating senescent cells also show beneficial effects.
Human observational data supports the SASP-aging connection:
- Circulating levels of SASP factors like IL-6 and IL-8 increase with age and correlate with frailty, disability, and mortality risk.
- Higher hs-CRP levels predict cardiovascular events and all-cause mortality independent of traditional risk factors.
- Tissue analysis from aged humans shows accumulation of senescent cells expressing SASP markers in adipose tissue, liver, and vascular endothelium.
The translational evidence is more limited but growing. Early-phase clinical trials of senolytic drugs like dasatinib plus quercetin have shown improvements in physical function and reductions in inflammatory markers in patients with idiopathic pulmonary fibrosis and diabetic kidney disease. However, these studies are small and short-term, and whether senolytic interventions extend human lifespan remains unknown. The field is still determining optimal dosing strategies, identifying which patient populations benefit most, and understanding potential risks of eliminating senescent cells that may serve beneficial functions in wound healing and tissue repair.
An important caveat is that not all senescent cells are harmful. In acute settings like wound healing and tumor suppression, cellular senescence and the SASP serve protective functions by preventing damaged cells from proliferating and recruiting immune cells to clear threats. The problem arises when senescent cells accumulate chronically and the SASP becomes a persistent source of tissue-damaging inflammation. This context-dependence means that blanket elimination of all senescent cells may not be optimal, and more targeted approaches that selectively remove pathological senescent cells while preserving beneficial ones may be necessary.
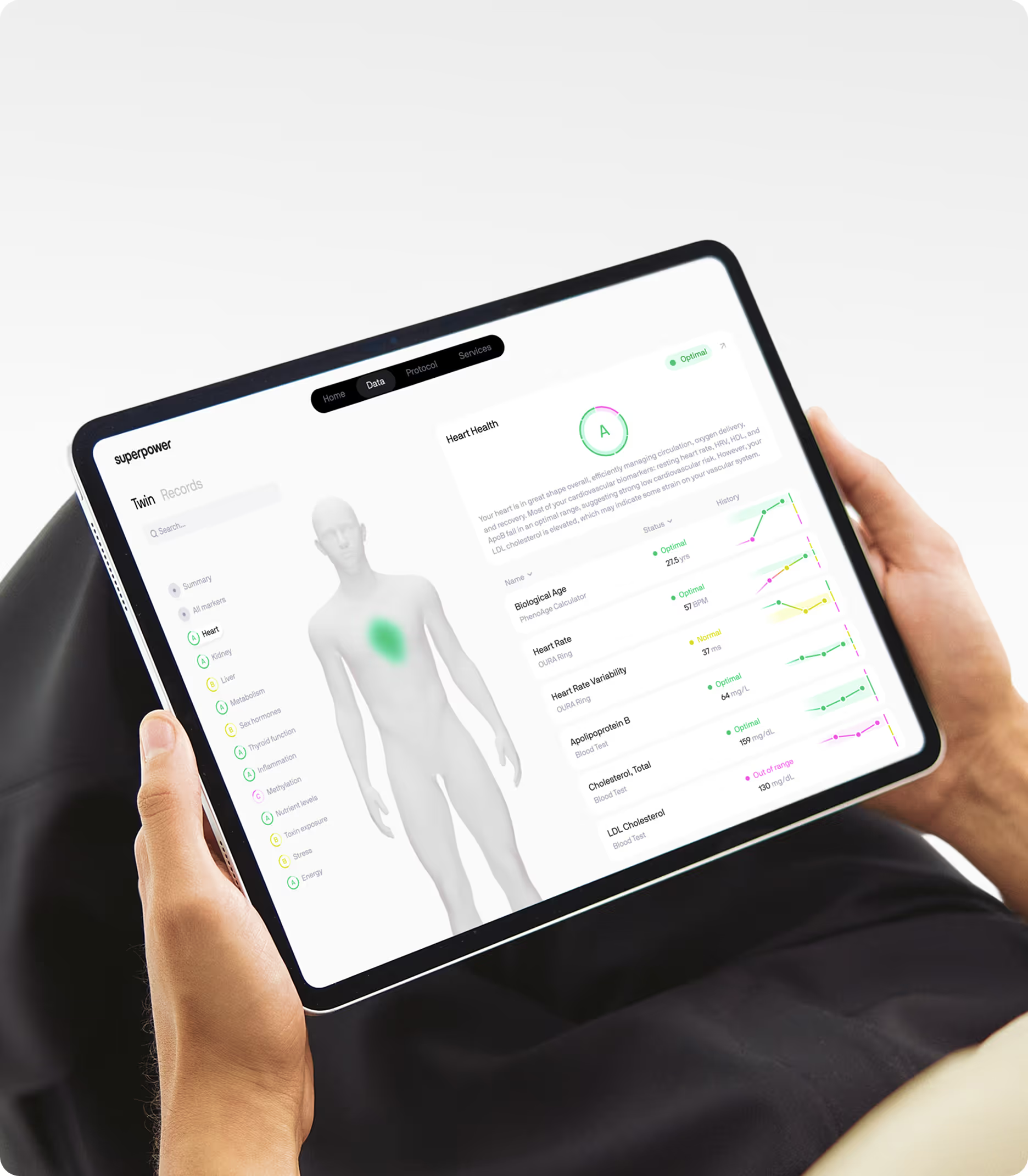
Measuring SASP-driven inflammation and cellular aging
Understanding your personal burden of SASP-driven inflammation requires measuring both systemic inflammatory markers and metabolic indicators that reflect the downstream consequences of chronic SASP exposure. While direct measurement of senescent cell burden in tissues requires invasive biopsies, blood-based biomarkers provide accessible proxies for SASP activity and its systemic effects.
Key markers include:
- High-sensitivity C-reactive protein reflects cumulative inflammatory burden from SASP-producing senescent cells (values below 1.0 mg/L indicate low risk, above 3.0 mg/L suggest high inflammatory burden).
- Erythrocyte sedimentation rate measures how quickly red blood cells settle in response to inflammatory proteins.
- Neutrophil-to-lymphocyte ratio and systemic immune-inflammation index provide composite measures of inflammatory status that correlate with biological aging.
- Fasting insulin and hemoglobin A1c reflect insulin resistance driven by SASP factors like IL-6.
- Triglyceride-glucose index provides an integrated measure of metabolic dysfunction that correlates with senescent cell burden.
- Apolipoprotein B and lipoprotein(a) reflect cardiovascular risk amplified by chronic inflammation.
Tracking these markers longitudinally provides more information than single measurements. Rising inflammatory markers over time suggest accumulating senescent cell burden and increasing SASP activity, while declining values indicate successful interventions. The rate of change in these biomarkers may be more predictive of biological aging rate than absolute values at any single timepoint.
Targeting the SASP for healthier aging
The senescence-associated secretory phenotype represents one of the most actionable targets in longevity science because it connects cellular aging to systemic inflammation and tissue dysfunction through measurable, modifiable pathways. Understanding your inflammatory and metabolic baseline through comprehensive biomarker testing allows you to track whether interventions are successfully reducing SASP-driven damage.
Superpower's Baseline Blood Panel measures the core inflammatory and metabolic markers that reflect SASP activity, including hs-CRP, complete blood count with differential, fasting glucose, insulin, hemoglobin A1c, and comprehensive lipid panel with apolipoprotein B. For those seeking deeper insight into inflammatory status, the Advanced Blood Panel adds markers like erythrocyte sedimentation rate and advanced lipid particle analysis that provide additional resolution on cardiovascular risk driven by chronic inflammation. These panels give you the data needed to establish your baseline inflammatory burden and track changes over time as you implement interventions targeting cellular senescence and the SASP.



FAQs
The SASP is the secretory profile that senescent cells develop after permanently exiting the cell cycle. Rather than dying quietly, senescent cells continuously release pro-inflammatory cytokines, chemokines, growth factors, proteases, and extracellular matrix remodeling proteins into their surrounding environment. This secretory activity transforms senescent cells from passive bystanders into active drivers of tissue dysfunction and chronic inflammation throughout the body.
The most prominent SASP components include interleukin-6 (IL-6) and interleukin-8 (IL-8), which are markedly elevated after DNA damage and oncogene-induced senescence. Additional components include IL-1α, IL-1β, tumor necrosis factor-alpha (TNF-α), monocyte chemoattractant protein-1 (MCP-1), matrix metalloproteinases (MMPs) that degrade the extracellular matrix, and vascular endothelial growth factor (VEGF). The exact composition varies based on cell type and the trigger that induced senescence.
SASP factors spread damage through several mechanisms. IL-1α and other cytokines can induce senescence in neighboring healthy cells through paracrine signaling, a process called bystander senescence. Matrix metalloproteinases degrade the extracellular matrix and disrupt tissue architecture. Recruited immune cells amplify local inflammation, and growth factors alter differentiation signals in nearby stem cells, reducing regenerative capacity across entire tissue compartments from a relatively small number of senescent cells.
IL-6 and IL-8 act in an autocrine manner, binding to receptors on the same senescent cells that produced them and strengthening the senescence growth arrest through NF-κB and mTOR signaling. Elevated mTOR in senescent cells drives ongoing translation of SASP factors. Cell-surface IL-1α acts as a master regulator, activating NF-κB to sustain downstream SASP expression. This self-reinforcing loop means senescent cells actively maintain their dysfunctional state rather than progressing to apoptosis.
SASP factors like IL-6 induce insulin resistance in surrounding tissues, creating metabolic dysfunction that compounds with age. The chronic inflammatory environment generated by SASP promotes atherosclerotic plaque instability, endothelial dysfunction, and cardiovascular events. In adipose tissue, SASP-secreting senescent cells drive systemic inflammation that contributes to type 2 diabetes progression. The same inflammatory cascade impairs neuronal function in the brain, linking senescent cell accumulation to neurodegeneration and cognitive decline.
The severity of an individual's SASP depends on immune system competence, as natural killer cells and macrophages normally clear senescent cells before they accumulate. Genetic polymorphisms in IL-6, TNF-α, and NF-κB pathway genes influence baseline inflammatory tone and SASP magnitude. Cumulative UV radiation, air pollution, and tobacco exposure accelerate senescent cell accumulation. Insulin resistance, elevated fasting insulin, declining sex hormones, and immunosenescence all enhance SASP intensity by increasing oxidative stress and reducing clearance efficiency.
References
- Wang, B., Han, J., Elisseeff, J. H., & Demaria, M. (2024). The senescence-associated secretory phenotype and its physiological and pathological implications. Nature reviews. Molecular cell biology, 25(12), 958-978. https://doi.org/10.1038/s41580-024-00727-x
- Coppé, J. P., Desprez, P. Y., Krtolica, A., & Campisi, J. (2010). The senescence-associated secretory phenotype: the dark side of tumor suppression. Annual review of pathology, 5, 99-118. https://doi.org/10.1146/annurev-pathol-121808-102144
- Tchkonia, T., Zhu, Y., van Deursen, J., Campisi, J., & Kirkland, J. L. (2013). Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. The Journal of clinical investigation, 123(3), 966-72. https://doi.org/10.1172/JCI64098
- Ohtani, N. (2022). The roles and mechanisms of senescence-associated secretory phenotype (SASP): can it be controlled by senolysis?. Inflammation and regeneration, 42(1), 11. https://doi.org/10.1186/s41232-022-00197-8
- National Institute on Aging. (n.d.). Mitochondrial DNA release drives cellular senescence and inflammation in mice. https://nia.nih.gov/news/mitochondrial-dna-release-drives-cellular-senescence-and-inflammation-mice

























.avif)
.svg)
.svg)


