.svg)


.svg)




How chronic stress drives biological aging at the cellular level
Chronic stress aging begins with the hypothalamic-pituitary-adrenal axis. When you encounter a stressor, your brain signals the adrenal glands to release cortisol. In acute situations, this response is adaptive: it mobilizes glucose, sharpens focus, and temporarily suppresses non-essential functions like digestion and reproduction. But when stress becomes chronic, cortisol remains elevated for weeks, months, or years. This sustained elevation shifts cortisol from a protective signal to a catabolic force that degrades muscle, bone, and immune function while promoting fat accumulation, particularly visceral fat.
The relationship between cortisol and biological age extends beyond metabolic disruption. Elevated cortisol directly alters gene expression through glucocorticoid receptors, which bind to hormone-sensitive regions of the genome and modify DNA methylation patterns. These epigenetic changes accumulate over time and are captured by epigenetic clocks like GrimAge and DunedinPACE, which measure biological age independently of chronological age. Studies show that individuals with high lifetime stress exposure exhibit accelerated epigenetic aging, meaning their cells appear older than their actual age would predict.
Cortisol also impairs cellular maintenance systems. It suppresses autophagy, the process by which cells clear damaged proteins and organelles. When autophagy is inhibited, misfolded proteins accumulate, mitochondrial function declines, and oxidative stress increases. This creates a feedback loop: oxidative stress damages cellular components, which in turn generate more reactive oxygen species, further overwhelming the cell's repair capacity. Over time, this contributes to loss of proteostasis, one of the primary hallmarks of aging.
Where chronic stress intersects with the hallmarks of aging
Chronic stress aging directly implicates several hallmarks of aging, particularly inflammation, cellular senescence, and genomic instability. Sustained cortisol elevation drives chronic low-grade inflammation, often termed inflammaging. Cortisol paradoxically promotes inflammation when chronically elevated because prolonged exposure leads to glucocorticoid receptor resistance. Immune cells become less responsive to cortisol's anti-inflammatory signals, allowing pro-inflammatory cytokines like interleukin-6 and tumor necrosis factor-alpha to remain elevated.
Inflammaging feeds into cellular senescence. Senescent cells are cells that have stopped dividing but remain metabolically active, secreting inflammatory molecules that damage neighboring cells. Chronic stress increases the accumulation of senescent cells through multiple mechanisms:
- Oxidative stress-induced DNA damage triggers cellular senescence pathways that prevent damaged cells from dividing.
- Telomere dysfunction activates DNA damage responses that force cells into permanent growth arrest.
- Mitochondrial impairment generates reactive oxygen species that accelerate cellular aging and senescence entry.
Once senescent cells accumulate, they amplify inflammation through the senescence-associated secretory phenotype, creating a self-reinforcing cycle that accelerates tissue aging.
Telomere attrition is another hallmark directly affected by chronic stress. Telomeres are protective DNA sequences at the ends of chromosomes that shorten with each cell division. When telomeres become critically short, cells enter senescence or die. Research shows that individuals with high perceived stress and elevated cortisol have shorter telomeres than age-matched controls. The mechanism involves both direct cortisol effects on telomerase activity and indirect effects through oxidative stress and inflammation, which accelerate the rate of telomere shortening beyond normal replicative aging.
Epigenetic alterations represent another intersection. Chronic stress modifies DNA methylation patterns in ways that silence protective genes and activate pro-aging pathways. These changes are not random; they cluster in regions associated with immune function, metabolic regulation, and stress response. The result is a biological age that diverges from chronological age, with stressed individuals showing epigenetic profiles more typical of older adults.
Mitochondrial dysfunction and metabolic dysregulation
Chronic stress impairs mitochondrial function through cortisol-mediated suppression of mitochondrial biogenesis and increased production of reactive oxygen species. Damaged mitochondria produce less ATP and more oxidative byproducts, which damage cellular membranes, proteins, and DNA. This mitochondrial dysfunction contributes to fatigue, reduced exercise capacity, and accelerated aging of metabolically active tissues like muscle and brain.
Deregulated nutrient sensing
Cortisol dysregulates nutrient-sensing pathways, particularly insulin signaling and mTOR activity. Chronic cortisol elevation promotes insulin resistance, which impairs glucose uptake and increases circulating glucose and insulin. Elevated insulin and glucose activate mTOR, a pathway that promotes growth and inhibits autophagy. While mTOR activation is necessary for tissue repair and growth, chronic activation accelerates aging by preventing cellular cleanup and promoting senescence.
What drives chronic stress and cortisol dysregulation
The inputs that drive chronic stress aging are both external and internal. Psychosocial stressors like job insecurity, caregiving burden, social isolation, and financial instability are among the most potent drivers of sustained cortisol elevation. These stressors activate the HPA axis repeatedly, preventing the system from returning to baseline. Over time, this leads to HPA axis dysregulation, characterized by either persistently elevated cortisol or a blunted cortisol response, both of which are associated with accelerated aging.
Sleep deprivation amplifies stress-induced aging. Sleep is when the body clears cortisol, consolidates memory, and activates repair processes. Chronic sleep restriction prevents cortisol from returning to baseline overnight, leading to cumulative elevation. Poor sleep also impairs the glymphatic system (which clears metabolic waste from the brain) and reduces growth hormone secretion, which is essential for tissue repair. The combination of elevated cortisol and impaired repair accelerates epigenetic aging and increases inflammation.
Physical inactivity worsens the effects of chronic stress. Exercise is one of the most potent modulators of the stress response:
- Muscle contraction increases brain-derived neurotrophic factor, which protects neurons from cortisol-induced damage.
- Regular movement promotes mitochondrial biogenesis, which counteracts stress-induced mitochondrial dysfunction.
- Physical activity enhances insulin sensitivity, reducing the metabolic burden of chronic cortisol elevation.
Sedentary individuals lack these protective adaptations, making them more vulnerable to the aging effects of chronic stress.
Dietary patterns influence stress resilience. Diets high in refined carbohydrates and low in antioxidants exacerbate oxidative stress and inflammation, amplifying the cellular damage caused by cortisol. Conversely, diets rich in polyphenols, omega-3 fatty acids, and fiber support antioxidant defenses and reduce systemic inflammation. Magnesium deficiency, common in Western diets, impairs HPA axis regulation and increases cortisol reactivity to stress.
Early life adversity programs the stress response for life. Individuals exposed to trauma, neglect, or chronic stress during childhood show persistently elevated cortisol reactivity and accelerated epigenetic aging in adulthood. This reflects epigenetic programming of the HPA axis during critical developmental windows, resulting in a stress response that is hyperreactive and slower to recover. This allostatic load accumulates over decades, contributing to earlier onset of age-related diseases.
Why stress resilience varies between individuals
Not everyone exposed to chronic stress ages at the same rate. Genetic variation in glucocorticoid receptor sensitivity determines how strongly cortisol affects target tissues. Individuals with certain polymorphisms in the glucocorticoid receptor gene show greater cortisol-induced inflammation and faster epigenetic aging in response to the same stressor. Similarly, genetic variants in genes encoding antioxidant enzymes like superoxide dismutase influence how effectively cells neutralize the oxidative stress generated by chronic cortisol elevation.
Baseline biological age matters. Two individuals of the same chronological age can have vastly different biological ages as measured by epigenetic clocks. Those starting with a younger biological age have more reserve capacity to buffer the effects of stress. Conversely, individuals with accelerated baseline aging due to prior exposures, poor metabolic health, or genetic predisposition show steeper declines when exposed to chronic stress.
Psychological resilience modulates the physiological stress response. Individuals with strong social support, high perceived control, and effective coping strategies show lower cortisol reactivity to stressors and faster cortisol recovery. This is not merely psychological; social connection directly influences immune function and inflammation through neural pathways that link the brain to the immune system. Loneliness and social isolation, by contrast, amplify cortisol responses and accelerate epigenetic aging independently of other risk factors.
Hormonal context influences stress vulnerability. Women show greater cortisol reactivity during certain phases of the menstrual cycle and during perimenopause, when estrogen fluctuations destabilize HPA axis regulation. Men with low testosterone show exaggerated cortisol responses and impaired recovery. These hormonal differences contribute to sex-specific patterns of stress-related aging and disease risk.
What the evidence actually shows on stress and aging
The link between chronic stress and accelerated biological aging is supported by multiple lines of human evidence. Longitudinal studies using epigenetic clocks show that individuals with high cumulative stress exposure exhibit faster rates of epigenetic age acceleration. The MIDUS study, which followed thousands of adults over decades, found that childhood adversity and chronic adult stress were associated with biological age acceleration of several years, even after controlling for health behaviors and socioeconomic status.
Telomere studies provide converging evidence. Meta-analyses show that chronic psychological stress is associated with shorter telomeres across diverse populations. The effect size is modest but consistent, with high-stress individuals showing telomeres approximately 200-300 base pairs shorter than low-stress controls. This translates to roughly 3-6 years of accelerated cellular aging. Importantly, the relationship is dose-dependent: greater cumulative stress exposure predicts shorter telomeres.
Intervention studies demonstrate reversibility:
- Mindfulness-based stress reduction programs lasting 8 weeks reduce cortisol levels and decrease inflammatory markers like high-sensitivity C-reactive protein.
- Some meditation interventions increase telomerase activity, the enzyme responsible for maintaining telomere length.
- Controlled trials comparing forest bathing to urban walking demonstrate that time in natural environments reduces cortisol, lowers blood pressure, and decreases sympathetic nervous system activity.
- Structured social engagement programs reduce loneliness and improve inflammatory markers in isolated individuals.
While these studies are relatively short-term and do not yet prove lifespan extension, they establish that stress-reduction interventions can favorably shift biomarkers associated with aging.
Social connection interventions show promise but are harder to study rigorously. Observational data consistently link strong social ties to lower mortality risk and slower biological aging. However, randomized trials of social interventions are limited. The available evidence suggests that structured social engagement programs reduce loneliness and improve inflammatory markers, but whether these effects translate to measurable changes in biological age remains an open question.
Measuring stress-related aging with biomarkers
Tracking how chronic stress affects your aging trajectory requires measuring markers that reflect both the stress response and its downstream effects on cellular aging. Cortisol is the most direct measure of HPA axis activity. Morning cortisol reflects baseline HPA axis tone, while the cortisol awakening response captures HPA axis reactivity. Persistently elevated morning cortisol or a blunted awakening response both indicate HPA axis dysregulation.
Inflammatory markers provide insight into stress-induced inflammaging:
- High-sensitivity C-reactive protein is the most widely used marker of systemic inflammation and predicts cardiovascular disease and mortality risk.
- Interleukin-6 and tumor necrosis factor-alpha are more specific markers of chronic inflammation but are less commonly measured in clinical practice.
- Elevated inflammatory markers in the context of chronic stress suggest that stress is driving systemic inflammation.
Metabolic markers reflect cortisol's effects on glucose and lipid metabolism. Fasting insulin, hemoglobin A1c, and the triglyceride-to-HDL ratio capture insulin resistance and metabolic dysfunction driven by chronic cortisol elevation. Apolipoprotein B and lipoprotein(a) provide additional cardiovascular risk stratification, as chronic stress accelerates atherosclerosis through both metabolic and inflammatory pathways.
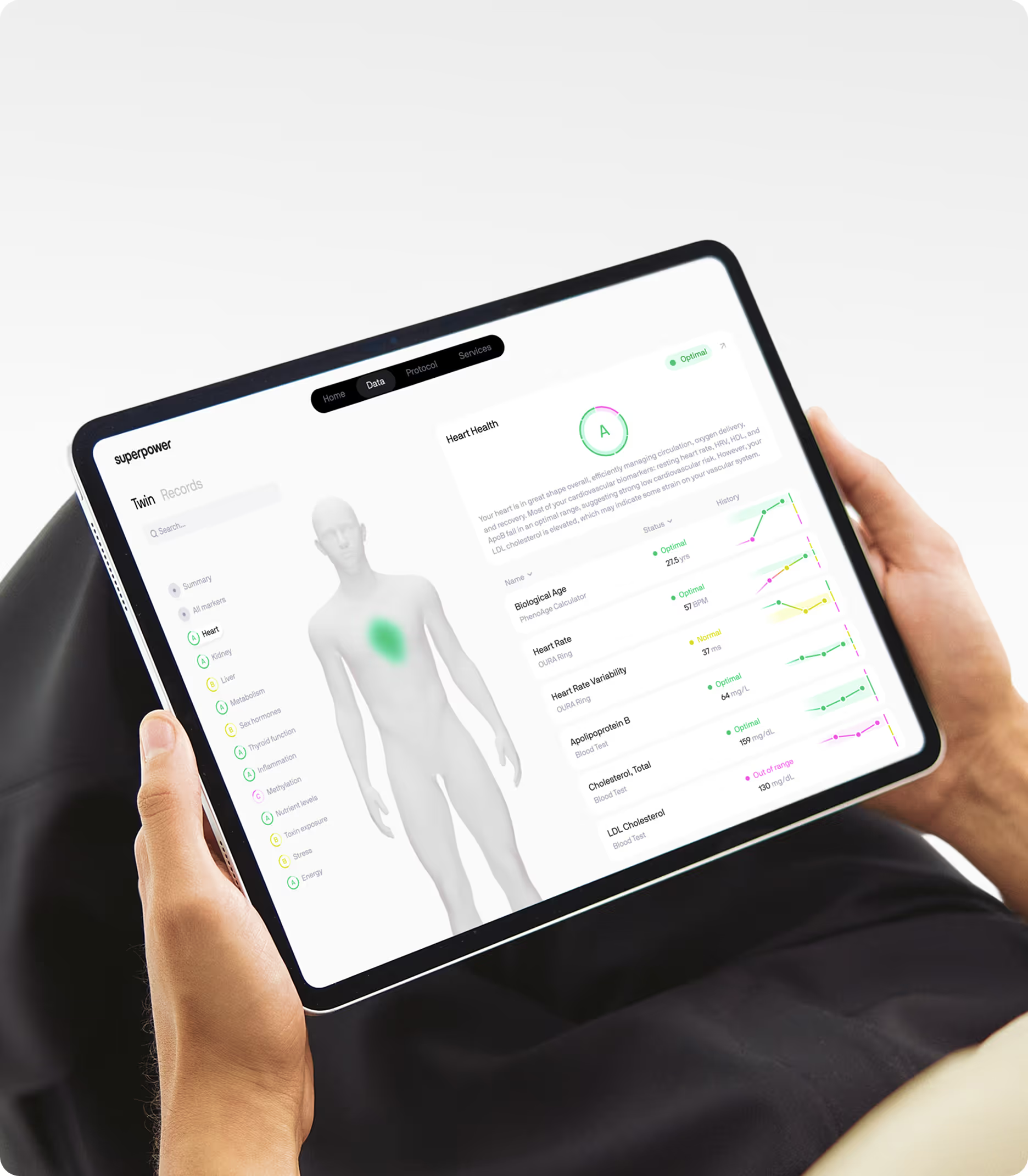
Epigenetic age testing offers a direct measure of biological aging. Clocks like GrimAge and DunedinPACE integrate information from hundreds of DNA methylation sites to estimate biological age and pace of aging. These tests are increasingly available commercially and provide a snapshot of how stress and other exposures have affected your aging trajectory. Serial testing over time can reveal whether interventions are slowing your pace of aging.
Tracking these markers longitudinally provides more information than a single measurement. A rising cortisol-to-DHEA-S ratio, increasing inflammatory markers, or accelerating epigenetic age all signal that chronic stress is driving biological aging. Conversely, stable or improving markers suggest that stress management interventions are working.
Building a data-driven approach to stress and aging
Understanding how chronic stress accelerates aging is only useful if you can measure it in your own body. Superpower's 100+ biomarker panel includes cortisol, high-sensitivity C-reactive protein, fasting insulin, hemoglobin A1c, and metabolic markers that reveal how stress is affecting your metabolic health and inflammatory tone. These are the markers that standard annual bloodwork typically misses, yet they provide the clearest picture of whether chronic stress is driving biological aging in your body. Measuring these markers over time lets you track whether stress-reduction interventions are shifting your biology in the right direction, giving you a real baseline for how you're aging at a cellular level.



FAQs
Chronic stress drives aging through sustained cortisol elevation that suppresses autophagy, impairs mitochondrial function, promotes DNA damage, and alters DNA methylation patterns via glucocorticoid receptors. These changes accumulate across hallmarks of aging including inflammation, cellular senescence, telomere attrition, and loss of proteostasis.
Elevated cortisol binds to glucocorticoid receptors and modifies DNA methylation patterns. These epigenetic changes accumulate over time and are captured by clocks like GrimAge and DunedinPACE. Studies show individuals with high lifetime stress exposure exhibit epigenetic profiles more typical of older adults than their chronological age would predict.
Chronic stress shortens telomeres through both direct and indirect mechanisms. Cortisol reduces telomerase activity—the enzyme that maintains telomere length—while oxidative stress and inflammation accelerate telomere attrition beyond normal replicative aging rates. Meta-analyses show high-stress individuals have telomeres roughly 200 to 300 base pairs shorter than low-stress controls.
Inflammaging is the chronic low-grade inflammation that accelerates aging. Chronic cortisol paradoxically promotes inflammation because prolonged exposure causes glucocorticoid receptor resistance—immune cells stop responding to cortisol's anti-inflammatory signals, leaving pro-inflammatory cytokines like IL-6 and TNF-alpha chronically elevated.
Yes. Mindfulness-based stress reduction lasting 8 weeks reduces cortisol and decreases hsCRP. Some meditation interventions increase telomerase activity. Forest bathing reduces cortisol versus urban walking in controlled trials. These studies are short-term but establish that stress-reduction interventions can favorably shift aging-associated biomarkers.
Genetic variation in glucocorticoid receptor sensitivity, baseline biological age, early life adversity, psychological resilience, and hormonal context all modulate stress-related aging. Individuals with strong social support and effective coping strategies show lower cortisol reactivity. Women face greater vulnerability during perimenopause due to estrogen-HPA axis interactions.
References
- Harvard Health Publishing. (2011). Understanding the stress response - Harvard Health. https://health.harvard.edu/healthy-aging-and-longevity/understanding-the-stress-response
- Mayo Clinic. (n.d.). Stress. https://mayoclinic.org/healthy-lifestyle/stress-management/in-depth/stress/art-20046037
- Lin, J., & Epel, E. (2022). Stress and telomere shortening: Insights from cellular mechanisms. Ageing research reviews, 73, 101507. https://doi.org/10.1016/j.arr.2021.101507
- Cleveland Clinic. (n.d.). What Does Cortisol Do?. https://my.clevelandclinic.org/health/articles/22187-cortisol
- Polsky, L. R., Rentscher, K. E., & Carroll, J. E. (2022). Stress-induced biological aging: A review and guide for research priorities. Brain, behavior, and immunity, 104, 97-109. https://doi.org/10.1016/j.bbi.2022.05.016
























.avif)
.svg)
.svg)


